| MitImpact id |
MI.987 |
MI.989 |
MI.988 |
| Chr |
chrM |
chrM |
chrM |
| Start |
8989 |
8989 |
8989 |
| Ref |
G |
G |
G |
| Alt |
A |
C |
T |
| Gene symbol |
MT-ATP6 |
MT-ATP6 |
MT-ATP6 |
| Extended annotation |
mitochondrially encoded ATP synthase membrane subunit 6 |
mitochondrially encoded ATP synthase membrane subunit 6 |
mitochondrially encoded ATP synthase membrane subunit 6 |
| Gene position |
463 |
463 |
463 |
| Gene start |
8527 |
8527 |
8527 |
| Gene end |
9207 |
9207 |
9207 |
| Gene strand |
+ |
+ |
+ |
| Codon substitution |
GCC/ACC |
GCC/CCC |
GCC/TCC |
| AA position |
155 |
155 |
155 |
| AA ref |
A |
A |
A |
| AA alt |
T |
P |
S |
| Functional effect general |
missense |
missense |
missense |
| Functional effect detailed |
missense |
missense |
missense |
| OMIM id |
516060 |
516060 |
516060 |
| HGVS |
NC_012920.1:g.8989G>A |
NC_012920.1:g.8989G>C |
NC_012920.1:g.8989G>T |
| HGNC id |
7414 |
7414 |
7414 |
| Respiratory Chain complex |
V |
V |
V |
| Ensembl gene id |
ENSG00000198899 |
ENSG00000198899 |
ENSG00000198899 |
| Ensembl transcript id |
ENST00000361899 |
ENST00000361899 |
ENST00000361899 |
| Ensembl protein id |
ENSP00000354632 |
ENSP00000354632 |
ENSP00000354632 |
| Uniprot id |
P00846 |
P00846 |
P00846 |
| Uniprot name |
ATP6_HUMAN |
ATP6_HUMAN |
ATP6_HUMAN |
| Ncbi gene id |
4508 |
4508 |
4508 |
| Ncbi protein id |
YP_003024031.1 |
YP_003024031.1 |
YP_003024031.1 |
| PhyloP 100V |
7.779 |
7.779 |
7.779 |
| PhyloP 470Way |
0.965 |
0.965 |
0.965 |
| PhastCons 100V |
1 |
1 |
1 |
| PhastCons 470Way |
0.086 |
0.086 |
0.086 |
| PolyPhen2 |
probably_damaging |
probably_damaging |
probably_damaging |
| PolyPhen2 score |
1.0 |
1.0 |
0.99 |
| SIFT |
neutral |
deleterious |
neutral |
| SIFT score |
0.79 |
0 |
0.29 |
| SIFT4G |
Damaging |
Damaging |
Damaging |
| SIFT4G score |
0.018 |
0.015 |
0.001 |
| VEST |
Neutral |
Neutral |
Neutral |
| VEST pvalue |
0.36 |
0.15 |
0.34 |
| VEST FDR |
0.65 |
0.65 |
0.65 |
| Mitoclass.1 |
neutral |
damaging |
neutral |
| SNPDryad |
Neutral |
Pathogenic |
Pathogenic |
| SNPDryad score |
0.8 |
0.97 |
0.95 |
| MutationTaster |
Disease |
Disease automatic |
Disease |
| MutationTaster score |
0.99806 |
0.999445 |
0.995532 |
| MutationTaster converted rankscore |
0.4447 |
0.4708 |
0.42735 |
| MutationTaster model |
simple_aae |
simple_aae |
simple_aae |
| MutationTaster AAE |
A155T |
A155P |
A155S |
| fathmm |
Tolerated |
Tolerated |
Tolerated |
| fathmm score |
4.31 |
3.82 |
4.43 |
| fathmm converted rankscore |
0.02412 |
0.03728 |
0.02158 |
| AlphaMissense |
likely_pathogenic |
likely_pathogenic |
ambiguous |
| AlphaMissense score |
0.6292 |
0.9255 |
0.3458 |
| CADD |
Deleterious |
Deleterious |
Deleterious |
| CADD score |
4.295187 |
3.858663 |
3.743465 |
| CADD phred |
24.0 |
23.5 |
23.3 |
| PROVEAN |
Damaging |
Damaging |
Damaging |
| PROVEAN score |
-3.22 |
-4.39 |
-2.51 |
| MutationAssessor |
low |
high |
medium |
| MutationAssessor score |
1.935 |
4.34 |
2.99 |
| EFIN SP |
Neutral |
Damaging |
Neutral |
| EFIN SP score |
0.822 |
0.588 |
0.664 |
| EFIN HD |
Neutral |
Neutral |
Neutral |
| EFIN HD score |
0.468 |
0.436 |
0.514 |
| MLC |
Neutral |
Neutral |
Neutral |
| MLC score |
0.06524232 |
0.06524232 |
0.06524232 |
| PANTHER score |
0.472 |
. |
. |
| PhD-SNP score |
0.708 |
. |
. |
| APOGEE1 |
Neutral |
Neutral |
Neutral |
| APOGEE1 score |
0.21 |
0.37 |
0.35 |
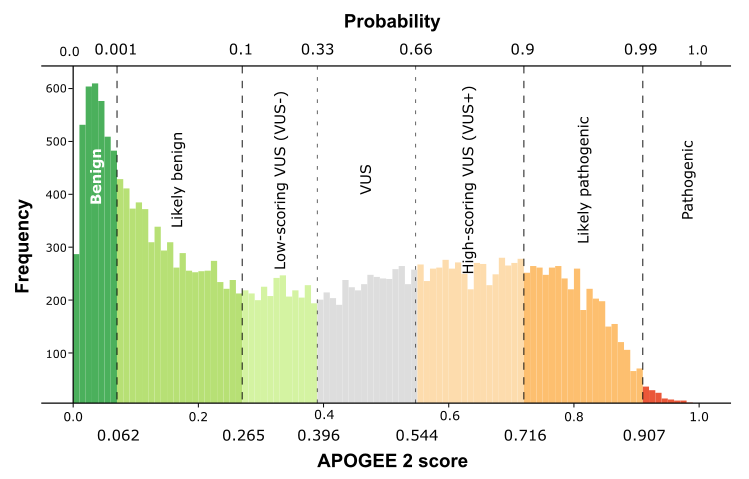
| APOGEE2 |
Likely-benign |
Likely-pathogenic |
Likely-benign |
| APOGEE2 score |
0.101801569016326 |
0.895475608124623 |
0.218675747790464 |
| CAROL |
deleterious |
deleterious |
deleterious |
| CAROL score |
1 |
1 |
0.99 |
| Condel |
neutral |
neutral |
neutral |
| Condel score |
0.4 |
0 |
0.15 |
| COVEC WMV |
neutral |
deleterious |
neutral |
| COVEC WMV score |
-2 |
6 |
-2 |
| MtoolBox |
deleterious |
deleterious |
deleterious |
| MtoolBox DS |
0.74 |
0.91 |
0.73 |
| DEOGEN2 |
Tolerated |
Tolerated |
Tolerated |
| DEOGEN2 score |
0.07066 |
0.299028 |
0.072529 |
| DEOGEN2 converted rankscore |
0.34015 |
0.67157 |
0.34477 |
| Meta-SNP |
Disease |
. |
. |
| Meta-SNP score |
0.51 |
. |
. |
| PolyPhen2 transf |
low impact |
low impact |
low impact |
| PolyPhen2 transf score |
-3.6 |
-3.6 |
-2.65 |
| SIFT_transf |
medium impact |
low impact |
medium impact |
| SIFT transf score |
0.62 |
-1.4 |
0.07 |
| MutationAssessor transf |
medium impact |
high impact |
medium impact |
| MutationAssessor transf score |
-0.01 |
2.28 |
0.05 |
| CHASM |
Neutral |
Neutral |
Neutral |
| CHASM pvalue |
0.75 |
0.76 |
0.86 |
| CHASM FDR |
0.9 |
0.9 |
0.9 |
| ClinVar id |
693046.0 |
155893.0 |
. |
| ClinVar Allele id |
681582.0 |
165642.0 |
. |
| ClinVar CLNDISDB |
MONDO:MONDO:0009723,MedGen:C0023264,OMIM:256000,Orphanet:506 |
MONDO:MONDO:0009723,MedGen:C0023264,OMIM:256000,Orphanet:506 |
. |
| ClinVar CLNDN |
Leigh_syndrome |
Leigh_syndrome |
. |
| ClinVar CLNSIG |
Likely_benign |
not_provided |
. |
| MITOMAP Disease Clinical info |
. |
NARP syndrome |
. |
| MITOMAP Disease Status |
. |
Reported |
. |
| MITOMAP Disease Hom/Het |
./. |
-/+ |
./. |
| MITOMAP General GenBank Freq |
0.0573% |
0.0% |
0.0% |
| MITOMAP General GenBank Seqs |
35 |
0 |
0 |
| MITOMAP General Curated refs |
9461455 |
23266623;30763462 |
. |
| MITOMAP Variant Class |
polymorphism |
disease |
polymorphism |
| gnomAD 3.1 AN |
56415.0 |
56432.0 |
. |
| gnomAD 3.1 AC Homo |
13.0 |
0.0 |
. |
| gnomAD 3.1 AF Hom |
0.000230435 |
0.0 |
. |
| gnomAD 3.1 AC Het |
6.0 |
0.0 |
. |
| gnomAD 3.1 AF Het |
0.000106355 |
0.0 |
. |
| gnomAD 3.1 filter |
PASS |
npg |
. |
| HelixMTdb AC Hom |
68.0 |
. |
1.0 |
| HelixMTdb AF Hom |
0.00034696888 |
. |
5.1024836e-06 |
| HelixMTdb AC Het |
31.0 |
. |
0.0 |
| HelixMTdb AF Het |
0.00015817699 |
. |
0.0 |
| HelixMTdb mean ARF |
0.30079 |
. |
. |
| HelixMTdb max ARF |
0.88406 |
. |
. |
| ToMMo 54KJPN AC |
23 |
. |
. |
| ToMMo 54KJPN AF |
0.000424 |
. |
. |
| ToMMo 54KJPN AN |
54302 |
. |
. |
| COSMIC 90 |
. |
. |
. |
| dbSNP 156 id |
rs587776444 |
rs587776444 |
. |